A few years ago, we were asked by Vietnam’s National Institute of Hygiene and Epidemiology to help analyze their influenza sequence data. The initial question on everyone’s mind was “where does Vietnam’s influenza come from?”, but it became clear pretty quickly that flu viruses can come from anywhere. The global phylogeography of influenza had already been described by a few large-scale sequence analyses (here, here, here, and here), showing that influenza A viruses circulate to and from all parts of the world. Even in the in the eight years of influenza sequence data that we had for Vietnam, it was clear that in any year, an introduction of influenza virus into the country could have come from a large number of other countries.
Like most previous studies, we tried to be careful to account for sampling bias in the influenza sequence database. Virus sequences are submitted to genbank by individual investigators, by WHO collaborating centers, by Ministries of Health, and other national or regional public health agencies. The end result is a clumped and eclectic collection of sequences. Hundreds of sequences from the Netherlands, but only two dozen from Belgium. From New Zealand, 355 influenza H3 sequences were submitted between 2003 and 2005, but only 38 in the three years that followed. When performing phylogeographic analyses on these sequences sets, it is important to know if some years or regions are oversampled or undersampled. To correct for the large variation in sample sizes, the standard solution is to subsample the data so that each region/country in each year has the same number of sequences. We started with all of the human influenza sequences in GenBank, and we downsampled so that each one of our 27 predefined regions had 12 sequences per year. This strategy solves for oversampled regions or years, but not for undersampled ones.
Using a parsimony method to reconstruct migration events, we were able to represent global migration patterns of H3 influenza, between 2001 and 2008, in the map below.
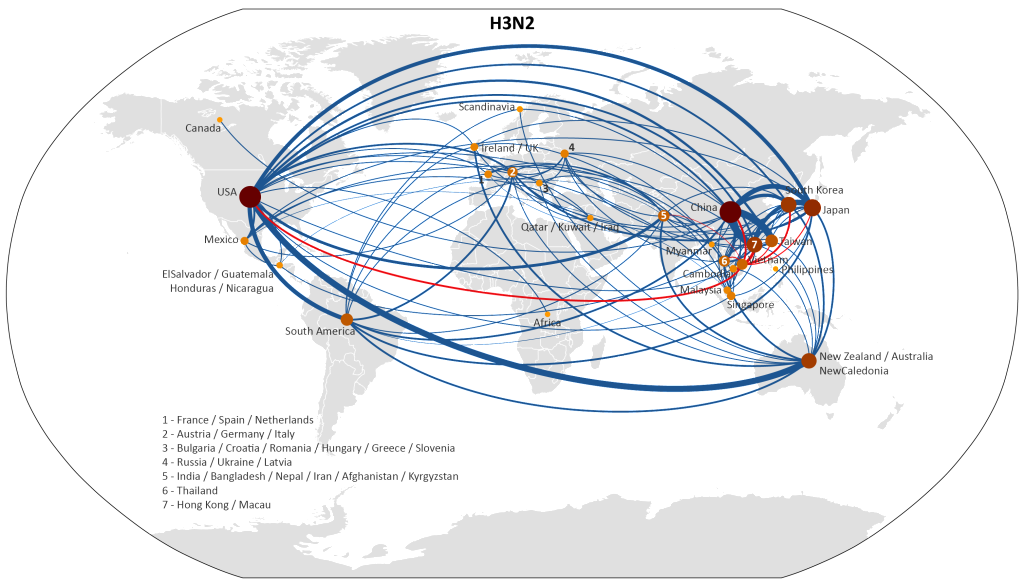
The red lines show migration events between Vietnam and other countries, and the width of each line corresponds to the number of migration events inferred between those countries. Click here for a full-size version of this map.
The more interesting finding, however, was the relationship between sample size and the number of migration events.
After subsampling, different region-year combinations still had different numbers of sequences, as there was no way to correct for undersampling. When plotting the number of migration events inferred for each region against the number of samples available by region, the result looked like this:
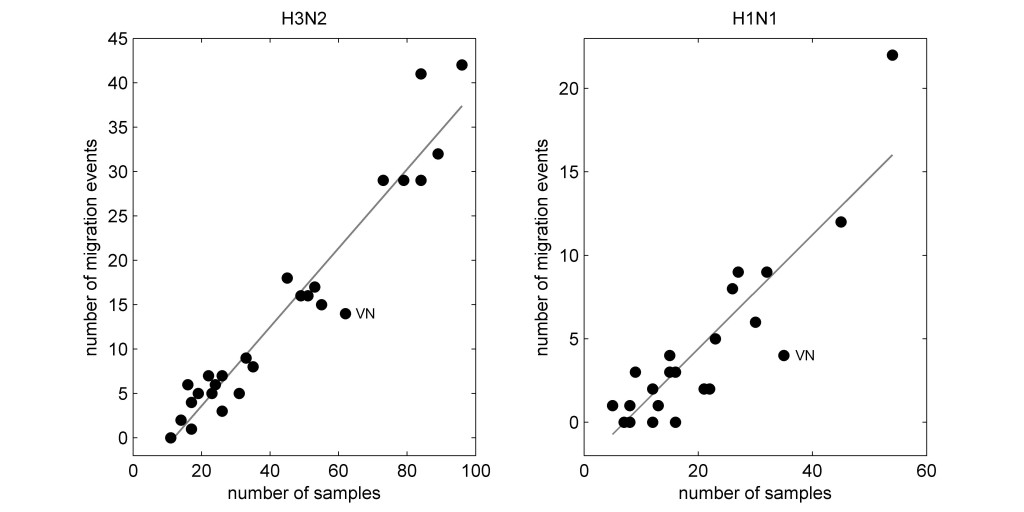
Thus, even in a subsampled data set, correction for oversampling still leaves us with a data set where some regions have more sequences than others, and the sample count for each region is closely correlated (positively) to the number of migration events inferred for that region.
Should we be subsampling in phylogeographic analyses? Should regions and countries be subsampled so that each one has the same number of sequences? We don’t know the answer to this question because, when a particular country is overrepresented or underrepresented in the influenza sequence database, we don’t know which way causality runs between sample count and migration importance. GenBank has very few influenza sequences from Ecuador, and therefore Ecuador would typically be inferred in phylogeographic analyses to be a global migration link of low importance. But causality can operate in the other direction. Perhaps Ecuador truly has low influenza prevalence — and thus a small number of sequences submitted to GenBank each year — and its low prevalence makes it a globally unimportant source of influenza viruses.
Having estimates of prevalence or attack rates by country would make geographically representative sampling possible. However, these measures are not collected in most countries, and certainly not on a year to year basis.
Our publication in Emerging Infectious Diseases can be found here.